
Linear prediction estimates the value of a point based on the values of adjacent points. This method can be used to replace corrupted values in an FID (Free Induction Decay). For example, in NMR experiments with fast relaxation rates, data collection must begin almost immediately following an RF pulse, even before the receiver and preamplifier can fully recover from ringing and saturation caused by the RF pulse. Consequently, several of the first datapoint values can be corrupted by instrumentation-induced noise. The effect of this noise on the spectrum can be profound because it often amounts to large fractions of the highest-amplitude signal values. Therefore the spectrum can improve noticeably when these corrupted point values are replaced with values estimated by linear prediction, where the estimated values are based on subsequent points of greater integrity.
A second application of linear prediction to NMR data is to extend an FID. This is useful for experiments in which data collection ceased before the signals completely decayed; that is, the FID is truncated. Here, the data values of the FID are used to estimate new data values that are appended to the end of the FID. Often the FID can be extended 20% or more by this means.
The Process1D/Linear Prediction/First Point Prediction menu item uses linear prediction to replace data values at the beginning of the FID, while the Process1D/Linear Prediction/Last Point (or general) Prediction menu item uses linear prediction to replace data values at the end of the FID and can also be used to extend the FID. The mathematical underpinnings of the linear prediction algorithm are discussed next, so that in the ensuing descriptions of the menu items, the settings in the control panels may be related to the mechanics of the algorithm.
Forward prediction is based on the assertion that the value of the nth data point Xn is determined from the values of K antecedent points:
| Eq. 1 |
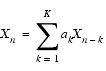
|
where ak are the linear prediction (LP) coefficients. These LP coefficients are determined from n-k values of Xn, cast in matrix notation as:
| Eq. 2 |
|
In general, the matrix C is over-determined; therefore, the solution to this equation is found by least-squares analysis, which is based on a singular value decomposition in order to ensure a solution when CTC is not invertible:
| Eq. 3 |
|
The matrices n, L, and u are obtained from the singular value decomposition of the matrix C. The matrix L is a diagonal matrix of singular values of the columns of n and u containing the right and left singular vectors.
With knowledge of the maximum number of signals in the FID, the number of singular values in the matrix L can be reduced by truncating the matrices n, L-1, and uT. The result is reduced noise in the predicted points (Makhoul 1978, Kumaresan and Tufts 1982, Barkhuijsen et al. 1985).
To assure that the predicted data points are well behaved; i.e., decaying, the technique of "root reflection" is often used. In this procedure, one constructs a complex polynomial defined by the LP coefficients. Then the roots are determined from this polynomial. For a noiseless FID, the roots should lie within the unit circle, which, of course, does not always hold for real data. To enforce proper behaviors, one reflects all the roots into the unit circle and reconstructs the polynomial from which a new set of LP coefficients are extracted (see e.g., Zhu and Bax 1993).
For backward linear prediction, you start with an analogous linear equation, as in Eq. 1:
| Eq. 4 |
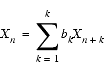
|
The mathematics of determining bk are similar to that used for forward prediction. However, you must pay attention when using root reflection that the roots in this case are reflected outside the unit circle.
Lastly, as pointed out by Zhu and Bax (1993), it is sometimes advantageous to perform both backward and forward predictions to obtain averaged LP coefficients. This procedure tends to be very good at suppressing noise and artifacts.
As mentioned earlier, linear prediction is accessed using Process1D/Linear Prediction/First Point Prediction or Process1D/Linear Prediction/Last Point (or general) Prediction. Choosing any of these menu items displays a control panel. The following three controls, which determine how the LP calculation is performed, are set in the control panels: Points to use, Number of coefficients, and Number of peaks. Points to use defines the number of points used to calculate the LP coefficients ak or bk. This corresponds to the value n in Eq. 1. The Number of coefficients corresponds to the value n-k. And Number of peaks is used to truncate the matrices n, L, and u (Eq. 3).
FELIX 2004 has several alternative autophasing methods for 1D spectra and one method for 2D and 3D spectra. The basic method and the method based on peak integration are used exclusively for 1D spectra. The method described in this section is based on PAMPAS (Dzakula 2000) or APSL (Heuer 1991) and is commonly used for 1D, 2D, and 3D spectra.
For a 1D spectrum in the workspace or a selected dimension of an ND spectrum, FELIX divides the spectrum into 10-40 segments of equal widths along the phasing dimension and tries to find a best sample peak per segment. The phase error of each individual sample peak is detected using the PAMPAS or APSL algorithm, and then the global zero- and first-order phase errors (represented by FELIX symbols phase0 and phase1) are obtained by linear regression analysis.
If solvent peaks or other noise peaks exist in the spectrum, it is important to exclude them from the phase detection process. FELIX allows you to define up to 10 excluded areas (using the aph exclude command). An excluded region is specified by the starting and ending datapoint numbers, and the dimension if it is an ND spectrum (which can be different from the phasing dimension). Any peaks that fall in the specified range along this dimension are not used as sample peaks.
For an ND spectrum, FELIX searches all the vectors along the phasing dimension and tries to find a best sample peak for each segment, without considering peaks in the excluded areas. Peaks are selected based on their width (must be wider than a user-defined threshold), height (the higher the better), and symmetry (the more symmetric the better). For a 1D spectrum, sample peaks are searched similarly in the only vector for each segment.
If more than two sample peaks are found for the whole spectrum, the phase errors are detected from each of them. Next, the global phase parameters (phase0 and phase1) are obtained by linear regression analysis. The phase errors detected are between 0� and 180�, which do not reflect the actual phase differences between them. The possible p-jumps between the individual phase errors are automatically compensated within the expected range of phase1. The default range of phase1 is approximately -720� to 720�. You can change the range of phase1 when using the aph command. See FELIX Command Language Reference for more details about using this command.
One approach to making assignments, developed by W�thrich and coworkers (W�thrich 1986), is the "sequential assignment strategy", which obtains sequence-specific resonance assignments (see Figure 1.).
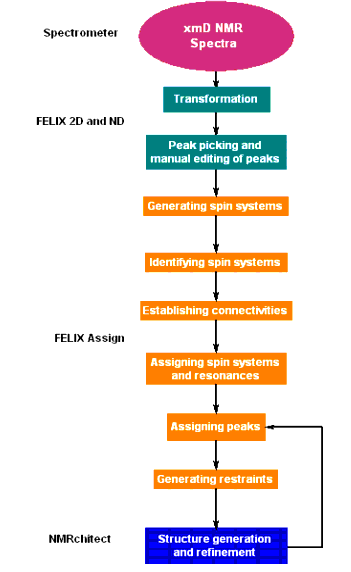
Figure 1. Sequential Assignment Strategy
The first step in this procedure is the delineation of individual spin systems, based on homonuclear 2D and/or 3D spectra or on heteronuclear spectra. If homonuclear 2D spectra are utilized, this spin-system delineation can be based on COSY type spectra (e.g., DQF-COSY or P-COSY). A spin system is understood to be a group of spins connected by scalar or through bond spin-spin couplings. This means that, for homonuclear spectra of a peptide or protein, one amino acid residue can contain one or more spin systems. However, due to the peptide bond (which yields a structure in which no non-labile hydrogens are three bonds apart in neighbor residues) spin systems do not comprise spins from more than one residue. Therefore, in a COSY-type spectrum, cross peaks are expected between scalarly coupled spins within individual residues.
For spin-system delineation, the TOCSY or HOHAHA type spectra can also be used, since the cross peaks arise only within individual spin systems. Ideally, TOCSY contains more cross peaks for the same spin system than COSY; thus, in complicated overlapping situations it is advantageous to combine the information from both types of spectra.
For spin-system delineation or detection it is possible to use two different methods: systematic searching (when all the spin systems in the spectrum are collected through systematically searching the spectrum for well-aligned peaks) and simulated annealing.
The complete delineation of spin systems is facilitated by the fact that the chemical shifts of the protons are in well-defined regions depending on structure (primary or secondary). The expectation values have been extracted for different protons in amino acid residues, based on analysis of assigned proteins (Gross 1988). You would expect to find most HN-Ha cross peaks at 6-12 ppm and 3-6 ppm. The chemical shift information and the pattern of cross peaks is necessary, but not sufficient by itself, for identification of spin systems with specific residue types, since neither all the chemical shifts nor all the cross peak patterns are unique.
To reduce ambiguities, additional homonuclear experiments can be performed (either NOESY or changing the solvent, pH, or temperature). In more difficult situations, heteronuclear experiments may also be added, if possible.
After spin-system delineation is complete, typically some spin systems remain that are not identified uniquely. Also, unique residue type identification is not always enough, since most biopolymers contain several copies of most residue types. These facts then necessitate the second step of sequence-specific resonance assignment, i.e., matching spin systems with specific residues. This is done through identification of NOE connectivities between spin systems corresponding to neighbor residues in the sequence.
Rigorous analysis of X-ray structures of globular proteins (Billeter et al. 1982) showed that several inter-residue distances are almost always small enough to yield cross peaks in a NOESY spectrum. These are, in particular: daN(i,i+1), dbN(i,i+1), and dNN(i,i+1). Usually daN(i,i+1) and dbN(i,i+1) are not short enough. Therefore, these distances can be used to tie together stretches of spin systems by analyzing the NOE interactions between protons of different spin systems. If such stretches can be matched with stretches of residue sequence, sequence-specific assignment is achieved with a high level of confidence.
Since NMR data tend to be incomplete or ill-resolved, a systematic search of perfect assignments yields only partial results. One has to tolerate a significant amount of imperfection and use loose criteria in order to find a probable assignment. Even then, an exhaustive inspection of all possible solutions might become impossible within reasonable computation times. Optimization methods such as simulated annealing then provide a powerful alternative; all constraints are used to define an energy for the system, which is minimized according to careful schemes. No general tolerance has to be adjusted. The minimizer spontaneously allows imperfections where (and only where) no better solution can be found with the available data (just as it does if peaks are missing from a TOCSY pattern, or if NOEs or even residues are missing from a sequential assignment) (Morelle et al. 1994a, 1994b).
The following steps were used by Eccles et al. (1991) for sequence-specific assignment of proteins based on homonuclear 2D spectra (DQF-COSY, TOCSY, and NOESY):
a. Start with the HN-Ha cross peak in DQF-COSY.
b. Identify all cross peaks belonging to this spin system using TOCSY (the alignment of COSY and TOCSY spectra may be rather poor-- 0.03 ppm).
c. Check all resulting groups against a database of 20 amino acid residues and score them.
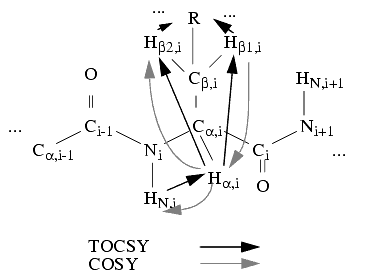
Figure 2. Homonuclear spin systems
a. A search can be carried out along the w2 direction at the Ha, Hb,... resonances of each spin system (with an allowed uncertainty of 0.01 ppm) to find the corresponding HN resonance of the sequential neighbor spin system. This can yield a list of NOE connectivities between the alpha, beta, and amide protons of each spin system and the amide proton of other spin systems.
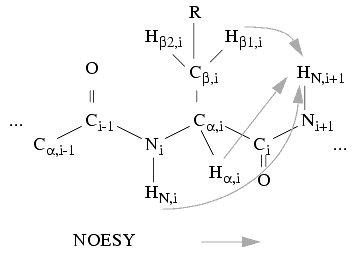
Figure 3. Sequential connections between spin systems
b. Uninterrupted pathways can be searched for, using the sequential NOEs and the amino acid sequence.
For spin-system delineation you can use (besides 2D spectra) homonuclear 3D spectra such as the 3D TOCSY-TOCSY spectrum (Kleywegt et al. 1993). The spin system can be connected sequentially via 3D TOCSY-NOE or 3D NOE-NOE spectra.
The more reliable and more efficient assignment strategy is based on higher dimensional heteronuclear spectra of singly or doubly labelled proteins or nucleic acids (some examples include Redfield et al. 1994, Hansen et al. 1994, Freund et al. 1994, Clubb et al. 1994, Anglister et al. 1994). Here, several methods of assigning singly or doubly labelled proteins are briefly described. The number of experiments that have been carried out on nucleic acids is still quite limited; for more detailed information on these types of experiments please see Farmer et al. 1993, Sklenar et al. 1993, Marino et al. 1994, Legault et al. 1994.
One of the first methods described in the literature used several combinations of double- and triple-resonance experiments to unravel the assignment for much bigger proteins than was possible earlier with homonuclear methods (Grzesiek et al. 1992). The steps involved can be summarized as follows:
a. Some peaks could have been missing from some spectra. For example, if water presaturation was used to measure HNCA (which might have been necessary) the HN protons, which were in fast exchange, would not give a signal when compared to HNCO.
b. The typical peak halfwidth is 45-55 Hz for Ha and 35-45 Hz for Ca, therefore the peak positions were prone to ambiguities.
c. Some spectra could have given extra peaks. For example, HNCA gave extra peaks for HN,i-Ni-Ca,i-1, but this can be used in conjunction with HN(CO)CA. Special care was taken in automated assignment.
For smaller proteins you can use fewer triple-resonance experiments and still achieve good sequential assignment of the backbone. In particular, you can use a combination of two 3D triple-resonance experiments (Grzesiek and Bax 1992), which correlates the Cb, Ca, N, and HN resonances of the ith and i-1th residues using CBCANH or HNCACB and CACB(CO)NH experiments. Then the residue types can be identified using a database of Ca and Cb chemical shifts (Grzesiek and Bax 1993).
You can also choose to apply 4D methods using these steps. Although they are usually limited by spectrometer time, the information content of two 4D spectra can be very large. Ideally, for example, you can use an HNNCAHA or HACANHN experiment and an HACA(CO)NHN experiment to obtain a complete list of Hai, Cai, Ni, HN,i, Ha,i-1, and Ca,i-1 resonances. From this list you can then construct a full sequential assignment of the backbone (Boucher et al. 1992a, 1992b, Constantine et al. 1993).
The Assign module includes several building blocks: the database, the visualization and analysis tools, and the automated algorithms.
The database consists of the data structures, which are built on the FELIX database management system. Using this Assign tool, you can keep all necessary information in a compact binary file, from which the specific entities (i.e., tables) can be exported in ASCII format. Also, FELIX provides spreadsheet capability, allowing you to view and, in certain cases, edit the tables.
The Assign database is structured in several layers. First you define a project entity, which is a container entity (i.e., it is at the top level and contains all information about the lower-level entities). The project entity, in which the assignment is carried out, then contains spectrum parameters (e.g., plot appearance, peaks, and spectra type description); spin-system information (patterns and frequencies, together with properties); sequence information (if any); the library of chemical shifts of residues; and various entities needed in the assignment procedure.
Table 1 lists the most important entities and symbols used in an Assign project:
Table 1| Entity | Variable name | Schema | Function | ||||
|---|---|---|---|---|---|---|---|
Pointers for each pattern-to-resonance list, neighbor item, and residue type probability item |
|||||||
reg:reslis1 |
|||||||
Contains a generic shift, a set of specific shifts and possible names for a given resonance for each item |
|||||||
Possible neighbor patterns for each given pattern with probabilities |
|||||||
reg:rasn 1 |
Real atom names and pseudoatoms of the current molecule (no coordinates) |
||||||
reg:fulseq 1 |
|||||||
reg:rseq1 |
|||||||
reg:resbuf1 |
|||||||
|
1No variables defined
|
|||||||
Visualization and analysis tools are provided by the graphical user interface (GUI). Functionality in the Assign module is organized into several submenus. Menu items dealing with repetitive visualization tasks (e.g., changing plot type, redefining plot limits and plot parameters) are easily accessible at all times through icons at the top of the FELIX window. In addition, several commands that help the assignment process are mapped to the right mouse button (drawing frequencies, picking and removing peaks,and displaying correlated cursors). Menu items specific to Assign include tools for tiling patterns in different spectra, overlaying contour plots, easy access to strip plots, and displaying frequency lists from different sources on different spectra.
When working with multiple 3D/4D data sets at once, quick access to different spectra as well as coordinated navigation through 2D planes of different views of different spectra is crucial for successful assignment. Therefore multiple spectra can be connected in one, two, or three dimensions, letting you define which axes from which views are to be changed together when plot limits are changed. There is also the option, as mentioned above, to overlay multiple spectra in the same window.
Many automated algorithms that facilitate the sequential assignment strategy are found within the Collect Prototype Patterns, Frequency Clipboard/Compare Frequencies, Residue Type/Score Residue Type, Residue Type/Match Residue Type, Neighbor/Find Neighbor Via..., Sequential/Systematic Search, Sequential/Simulated Annealing, and Peak Assign/Autoassign Peaks menu items.
The prototype-pattern detection menu items serve as spin-system detection tools. FELIX currently has several methods for automatically detecting spin systems:
The 2D homonuclear systematic search algorithm performs the steps shown in Figure 4..
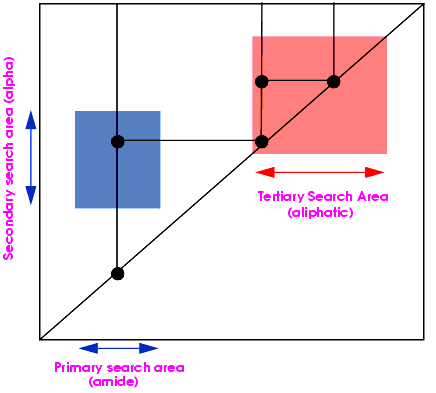
Figure 4. Homonuclear 2D systematic search
The 3D homonuclear spin-system detection algorithm can be used on a [J,J]-, a [J,NOE]-, or a [NOE,J]-type experiment and performs the steps in Figure 5..
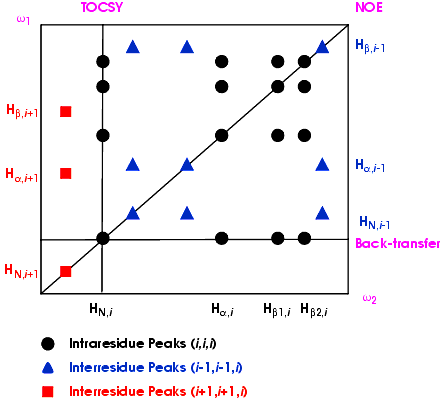
Figure 5. Typical components in the 2D w1-w2 plane of an AMX residue in a 3D TOCSY-NOE spectrum
The 2D simulated annealing detection algorithm uses these steps:
Assign implements several heteronuclear spin-system detection methods, which can be based on double- and triple-resonance experiments.
It is possible to use a 15N separated TOCSY experiment to collect spin systems starting on HN and N frequencies of the pseudodiagonal peaks (i.e., 15N-HN-HN peaks). The algorithm is similar to the 3D homonuclear one, but you need to specify the dimension that the root frequency (i.e., the HN) is in, and then all the frequencies are collected into spin systems that have peaks with the same HN and N frequencies. A method for collecting spin systems starting with 2D peaks is also implemented. For this method, you need to provide a 15N-1H HSQC and a 15N-HSQC-TOCSY spectra (certainly they can be HMQC, too).
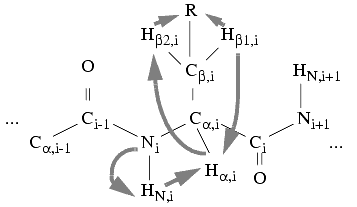
Figure 6. Spin systems in 15N-edited TOCSY experiments
Triple-resonance based spin-system detection algorithms are also provided in Assign. Particularly, you can use a combination of three 3D spectra-the HNCO, HN(CO)CA, and HNCA-and collect inter-residue spin systems or prototype patterns containing HN,i-Ni-Cai-Cai-1-Ci-1 frequencies. Then during promotion to patterns, an automated routine can find the sequential neighboring systems and store the results as neighbor probabilities.
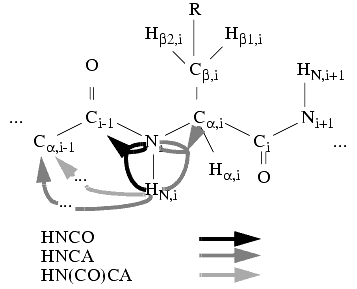
Figure 7. Spin systems in a typical triple-resonance 2D experiment
Similarly, you can use two triple-resonance 3D experiments-a CBCA(CO)NHN and a CBCANHN or HNNCACB-to collect prototype patterns containing HNi-Ni-Cai-Cbi-Cai-1-Cbi-1frequencies, and a similar promotion can be carried out. Now you can also use the 15N-1H HSQC spectrum to help collect the spin systems-mixing the information from heteronuclear double- and triple-resonance spectra.
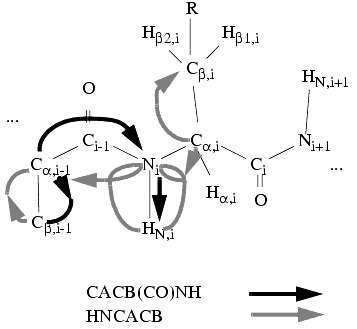
Figure 8. Spin-system detection in 3D triple-resonance spectra
In a similar method to that used in the previous example, you can collect spin systems in three triple-resonance experiments: HNCO, CBCANH, and CBCACO(N)H. This method results in spin systems containing: HNi-Ni-Cai-Cbi-Cai-1-Cbi-1-Ci-1.
It is possible to use an experiment to specifically find glycine spin systems (Wittekind 1993). A spin-system detection method is implemented in Assign for this, using the HNHA, CBCANH, and CBCA(CO)NH spectra.
Finally, you may use two 4D triple-resonance experiments-an HACA(CO)NHN and HACANHN-to automatically collect prototype patterns containing HNi-Ni-Cai-Hai-Cai-1-Hai-1 frequencies.
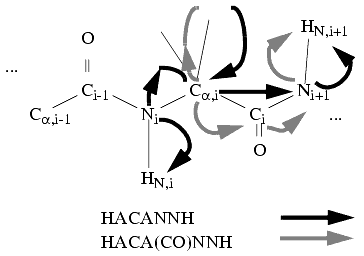
Figure 9. Spin-system detection in 4D triple-resonance spectra
A visual tool is provided to allow you to quickly find the previously detected spin systems in multidimensional spectra and to verify them visually.
The Frequency Clipboard/Compare Frequencies menu item checks a frequency list against a set of patterns or prototype patterns. The algorithm finds how many "fuzzy similarities" (Kleywegt et al. 1989) are present between the current collection of frequencies and the frequencies in each pattern or prototype pattern. You can use this to decide whether a new frequency list constitutes a new pattern.
The residue-type matching and scoring of patterns helps in assigning frequencies in patterns to specific residues. For each frequency in a pattern, the match-type action calculates a delta value based on the library expectation values and standard deviations collected for each residue type (delta = [actual_shift_of_freq - expectation_value]/standard_deviation). The score-type action automatically matches patterns and residue types. The algorithm tries to find a matching frequency in the pattern. The best-matching frequency may lie no more than a given number of standard deviations away from the expectation value. If at least a certain number of atoms can be matched, a score is computed.
The Neighbor/Find Neighbor Via... menu items serve to find potential neighbor patterns through a 2D NOESY, a 3D [J,NOE], a 3D [NOE,NOE], or a 15N-resolved NOESY spectrum. Therefore, this is the first step in sequence-specific assignment of patterns.
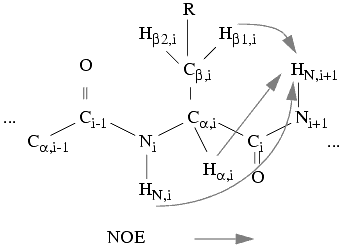
Figure 10. Neighboring spin-system detection based on NOE
The algorithm consists of these steps:
FELIX implements a triple-resonance-neighbor-finding algorithm in the Neighbor/Find Neighbor Via 3D/4D menu item. This action works on a triple-resonance experiment (e.g., HN(CO)CA or CBCA(CO)NHN) which contains cross peaks connecting neighboring residues. The algorithms for each pattern look through the spectrum and try to find peaks that connect this pattern to, for example, the i-1th residue. Then the sequential connectivity is set for the pattern that had the candidate frequency.
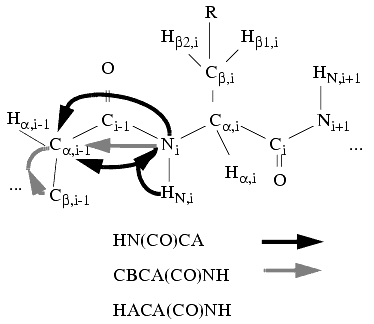
Figure 11. Neighboring spin-system detection based on triple- resonance spectra
The Sequential/Systematic Search and Sequential/Simulated Annealing menu items find potential matches of patterns with the sequence of an unbranched biopolymer. This can be done using systematic search methodology through a recursive matching procedure or using simulated annealing. In the latter case, the program tries to assign a set of patterns to the primary sequence (or a requested segment of it) of the molecule using optimization techniques rather than systematic searching. Each step in the simulated annealing routine is a permutation between the pool of patterns and a residue in the sequence (initially filled with empty dummy residues and, optionally, some previously assigned residues) or between two stretches of residues tentatively assigned in the primary sequence. The energy is computed from previously determined residue-fit scores and neighbor information. Eventually, the results converge to a solution of low energy, which is unique in well defined parts of the sequence. However, when the data are inadequate, several runs of the program might yield locally different solutions.
The Peak Assign/Autoassign Peaks menu item automatically assigns J or NOE peaks based on a set of assigned patterns, library, and molecule information. The patterns must have spectrum-specific shifts set for their resonances. The algorithm works as follows:
a. If one of the possible assignments has a distance connected to it that is shorter than the unambiguous cutoff and none of the others do, then this assignment is stored as a single assignment.
b. If more than one of the possible assignments has a shorter distance than the unambiguous cutoff, or none do, and multiple assignment is requested, then these are stored as multiple possible assignments.
NMR techniques for characterization of protein-ligand interactions are being widely used in rational drug design. As an example, the SAR by NMR strategy, devised by the Fesik group at Abbott Laboratories (Shuker et al. 1996; Hajduk et al. 1997), consists of the following general steps:
1. First, a library of small molecules is screened for binding to an 15N-labeled protein. If a molecule binds to the protein, it alters the local chemical environment and thereby cause changes in the chemical shifts of nuclei in the protein's binding site. Such changes are detected in HSQC spectra acquired in the presence and absence of added ligand.
2. Once initial ligands are identified, analogs are screened and binding constants are obtained in order to optimize the interactions with the protein.
3. Next, the 3D structure of the protein complexed with the ligands is obtained, and linked compounds are synthesized based on this structural information.
The Autoscreen module of FELIX automatically characterizes the protein-ligand interactions by identifying the changes between the reference and test spectra. With Autoscreen you can organize the project, process and score the 2D test spectra, and identify high-affinity ligands. For the SAR by NMR steps described above, Autoscreen offers help for the first two steps.
The core of Autoscreen consists a series of novel algorithms for tracing the shifting of peaks between the control and test spectra. In addition to the automatic peak tracing (APT) for high-throughput analysis, Autoscreen also provides an option for progressive peak tracing (PROPT) which is ideal for semi-automatic analysis of a series of titration spectra. In such a case, the shifting of the peaks is more reliably traced step-wise between the neighboring spectrum pairs following the titration curve.
A crucial phase in using the Autoscreen module is scoring of the test spectra against the control spectrum. Follow the steps below:
1. Pick peaks in the test spectrum.
2. Match test peaks to control peaks based on chemical shifts and peak shapes, and then score the experiment based on the displacements between the matched peak pairs.
3. Add penalties for the unmatched control and test peaks to the score.
The underlying principles are explained in the following sections.
The prerequisites for a test peak to be matched to a control peak are Eq. 5-Eq. 7.
| Eq. 5 |
|
| Eq. 6 |
|
| Eq. 7 |
|
where DH is the absolute displacement of the 1H chemical shift between the test peak and the control peak, maxDH the upper limit of this 1H displacement, DN the absolute displacement of the 15N chemical shift, maxDN the upper limit of the 15N displacement, S the shape similarity between the test peak and the control peak, and minS the lower limit of the shape similarity.
The test in Eq. 7 is applied when you choose to consider the shape of peaks. You can choose to define the shape similarity S as the product of the similarities of the peak widths and intensity:
| Eq. 8 |
|
where SwH, the similarity of peak widths along the 1H dimension, is calculated based on the width of the control peak ![]() and the width of the test peak
and the width of the test peak ![]() along the 1H dimension:
along the 1H dimension:
| Eq. 9 |
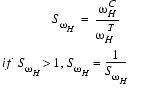
|
Analogously, ![]() , the similarity of peak widths along the 15N dimension, is calculated based on the width of the control peak
, the similarity of peak widths along the 15N dimension, is calculated based on the width of the control peak ![]() and the width of the test peak
and the width of the test peak ![]() along the 15N dimension:
along the 15N dimension:
| Eq. 10 |
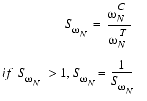
|
SI, the similarity of peak intensities, is calculated based on the peak heights of the control peak IC and of the test peak IT:
| Eq. 11 |
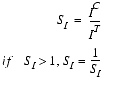
|
If a test peak is matched to a control peak, its contribution to the score of the experiment is:
| Eq. 12 |
|
where a and b are the weights of the 1H and 15N chemical shifts, respectively.
A control peak that does not match any test peak contributes a penalty c' (default is 0.6) to the score. The score of an experiment is hence the sum of the contributions of all matched and unmatched control peaks:
| Eq. 13 |
|
Sometimes some of the test peaks do not match any control peak. Although most unmatched test peaks are noise, any that are legitimate peaks can, if you choose to, contribute to the score such that:
| Eq. 14 |
|
where c" is the penalty for each unmatched test peak.
Normally, the contribution of peaks with too-small displacements are ignored, namely:
| Eq. 15 |
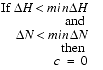
|
where minDH and minDN are the lower limits of the 1H and 15N displacements, respectively.
In practice you can change the weight of some control peaks to hide or highlight their contributions. In this way the reported score becomes:
| Eq. 16 |
|
where W is the weight of each control peak. Since test peaks are automatically picked on the fly, you cannot specify weights for them. Furthermore, although they contribute to the score, unmatched test peaks are not stored in the database as are other peaks.
Except in the simplest case, Eq. 5-Eq. 7 do not guarantee an unambiguous mapping of all test peaks to control peaks. Based on the assumptions that only a small portion of the peaks are displaced and that most peaks retain their locations and shapes in the test spectrum, Autoscreen assumes that the mapping that has a global-minimum value C as defined in Eq. 13 is the true mapping.
Peak picking in test spectra is done on the fly during scoring; i.e., you have no chance to optimize the picked peaks, hence the quality of the picked peaks is one of the key factors for successful scoring. Although most of the peak-picking parameters should remain identical to those used for peak picking in the control spectrum, the threshold may need to be adjusted for differences in sample concentrations.
Autoscreen first determines a peak-picking area, which is the minimum rectangle that includes all the control peaks plus margins of maxDH and maxDN in the D1 and D2 dimensions, respectively.
Autoscreen provides three ways of defining the threshold for peak picking: Automatic, Control, and Define. If the default setting (Automatic) is selected, an initial threshold is automatically calculated, and then the threshold is raised or lowered according to the following rules for each test spectrum:
1. If the number of test peaks is fewer than 1.1 times the number of control peaks, lower the threshold.
2. Otherwise, for each control peak, the test peaks that meet the criteria of Eq. 5-Eq. 7 are taken as candidate matches to the control peak. If over 10% of control peaks do not have a candidate match, lower the threshold.
3. Otherwise, if the number of test peaks is greater than 1.5 times the number of control peaks and each control peak has more than 3.0 candidate matches on average, raise the threshold.
No more than seven adjustment loops are performed.
If Control or Define is selected for the threshold method, the same threshold as that used for the control spectrum or a user-defined threshold is applied to all test spectra, respectively, and no automatic adjustment of thresholds is done.
For each control peak, the contributions of those test peaks that meet the criteria of Eq. 5-Eq. 7 are calculated using Eq. 12. These test peaks are sorted in ascending order of c, and the first four are retained as candidate matches. At this stage, similarities in peak heights (SI) are not used in Eq. 8, since the intensity level may be different between the control and test spectra.
To normalize the intensity level of the test spectrum, an intensity ratio IR is calculated as follows:
| Eq. 17 |
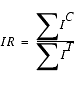
|
where ![]() IC is the sum of peak heights for all control peaks that have at least one candidate match, and
IC is the sum of peak heights for all control peaks that have at least one candidate match, and ![]() IT is the sum of peak heights of the first candidate matches to all control peaks. The peak heights of the test peaks are then normalized by multiplying by IR.
IT is the sum of peak heights of the first candidate matches to all control peaks. The peak heights of the test peaks are then normalized by multiplying by IR.
Next, the list of candidate matches is updated by including the peak intensity in
IC is the sum of peak heights for all control peaks that have at least one candidate match, and IT is the sum of peak heights of the first candidate matches to all control peaks. The peak heights of the test peaks are then normalized by multiplying by IR.
Next, the list of candidate matches is updated by including the peak intensity in Eq. 8.
The remaining task is to choose one or no test peak for each control peak, so that the sum of contributions (Eq. 13) is minimal. Some control peaks may not match any test peak, and such unmatched control peaks also contribute to the score.
Autoscreen provides two alternative methods for searching the mapping to find a global minimum for Eq. 13.
The first is a depth-first tree-search method. This deterministic method, in principle, should always find the true minimum, but in practice it may be too slow for complex spectra. Several heuristic methods are used to enhance its efficiency, so that, for normal spectra, satisfactory results are obtained in a few seconds. By default, the total CPU time is limited to 10 seconds although you can change this.
Another method is simulated annealing, a stochastic method that efficiently lowers the score (or energy) to or close to the global minimum. This method is recommended for very complicated spectra where tree searching does not give satisfactory results in a reasonable amount of CPU time.
There are many possible reasons for a control peak not to match any test peak. For example, if the upper limits for peak matching are set to too low a value, a peak that is shifted very far will not find its match. More often, two well separated peaks in the control spectrum become overlapped in the test spectrum. When this happens, only one test peak is picked, leading to one of the control peaks' being unmatched.
To unravel the overlapping test peaks in the latter case, Autoscreen fits such unmatched control peaks to the test spectrum using a local optimization method (see "Peaks/Optimize" on page 180). All unmatched control peaks, together with those neighboring control peaks that lie closer than four times their peak width to the unmatched control peaks, are fitted to the test spectrum with only the peak centers optimized. This process is iterated up to three times or until the change in the penalty is less than 5%. Next, the widths of these peaks are optimized for one iteration.
For each optimized peak, its equivalent peak height in the test spectrum is normalized using the intensity ratio, as for other test peaks. Then the optimized peak is matched to the original control peak to see if the conditions in Eq. 5-Eq. 7 are met.If this is successful, the contribution is calculated using Eq. 12. Otherwise, the control peak remains unmatched and its penalty is added to the score according to Eq. 13.
The fitting of unmatched control peaks to the test peaks is done only when the percentage of unmatched control peaks is below a certain threshold (default = 40%). If too many control peaks are not matched, this usually means that the spectrum is corrupted, and the fitting is not done. A corrupt spectrum usually has a score significantly higher than a normal spectrum, because of the contribution of unmatched control and test peaks.
Test peaks that are not matched to a control peak are usually noise, but they can also be real peaks corresponding to unwanted control peaks (e.g., the side-chain amide peaks are usually excluded from the control peaks), or to a control peak that was not noticed in the control spectrum because of peak overlap or other reasons. A legitimate test peak that was not observed in the control spectrum but that is picked in the test spectrum should contribute to the score.
Therefore, from all the unmatched test peaks, Autoscreen identifies legitimate test peaks based on their chemical shifts and peak shapes. If you choose to use the chemical-shift constraint, then only those peaks that are displaced less than maxDH and maxDN (in the 1H and 15N dimensions, respectively) with respect to their closest control peaks are considered. Next, the peak widths and heights of all the matched test peaks are analyzed statistically. At this point, an unmatched test peak is considered legitimate if its peak widths (wH and wN) and peak height (I) meet the following criteria:
| Eq. 18 |
|
| Eq. 19 |
|
| Eq. 20 |
|
where ![]() ,
, ![]() , and
, and ![]() are the average values of the peak width along the 1H dimension, the peak width along the 15N dimension, and the peak height, respectively; sH, sN, and sI are the standard deviations of the peak width along the 1H dimension, the peak width along the 15N dimension, and the peak height, respectively; and n is a user-defined coefficient (default value is 2.0).
are the average values of the peak width along the 1H dimension, the peak width along the 15N dimension, and the peak height, respectively; sH, sN, and sI are the standard deviations of the peak width along the 1H dimension, the peak width along the 15N dimension, and the peak height, respectively; and n is a user-defined coefficient (default value is 2.0).
By default, each unmatched test peak contributes a penalty of 0.20 to the score, though you can always change this parameter. This penalty is set smaller than that for an unmatched control peak, because you usually have a better chance to refine the control peaks based on the assignment. The test peaks, on the other hand, are automatically picked so are usually less reliable.
Presently, unmatched test peaks are only reported to you in the text window and in a log file, but not saved in the database like other peaks.
The Autoscreen/Setup Scoring menu item allows you to change or verify the parameters used for scoring. Table 2 correlates these parameter names with the symbols and terms mentioned in this chapter, for your convenience.
For more details about these parameters, please refer to "Processing and scoring menu items" on page 247.
Table 2| Parameter | Options | Default | Symbol or meaning | ||||
|---|---|---|---|---|---|---|---|
| Peak Displacement Limits (ppm) | |||||||
Use the same set of control peaks for all test spectra, or prompt for different set for each test spectrum. And the peak table entity name when Use One is selected. |
|||||||
Automatically pick test peaks, or prompt for a set of pre-picked test peaks for each test spectrum. And where to save the test peaks when Auto Pick is selected |
|||||||
minDH in Eq. 15 |
|||||||
maxDH in Eq. 5 |
|||||||
minDN in Eq. 15 |
|||||||
maxDN in Eq. 6 |
|||||||
| Scale Factors | |||||||
a in Eq. 12 |
|||||||
b in Eq. 12 |
|||||||
minS in Eq. 7 |
|||||||
Method to search for the global minimum. Options are Tree Search and Simulated Annealing. |
|||||||
Maximum CPU time (seconds) to spend searching the global minimum. Used only when Tree Search is selected. |
|||||||
| Unmatched Control Peaks | |||||||
Upper limit of unmatched control peaks, above which the spectrum is regarded as corrupt and no fitting is done. |
|||||||
Penalty for an unmatched control peak (c' in Eq. 13) |
|||||||
| Unmatched Test Peaks | |||||||
Penalty for an unmatched test peak (c" in Eq. 16) |
|||||||